The Newly Discovered Ring Structure of RAD52 Protein May Lead to New Treatments for BRCA-Deficient Cancers
The RAD52 protein is a coveted drug target for precision oncology as it is largely dispensable in healthy human cells but essential for survival of cancer cells that are deficient in DNA repair function, such as those with defective BRCA1 or BRCA2 genes. The discovery of the structural and functional information of RAD52 may help develop new, specific ways to inhibit this protein
Cancer cells thrive under conditions that would cripple normal cells. One of their survival tricks is dependency to backup DNA repair pathways when more commonly used processes fail. This vulnerability is the basis of synthetic lethality: a therapeutic strategy where the simultaneous inactivation of two genes (or two repair pathways) results in catastrophic cell death, even though the loss of either one alone is tolerated.
Among the next generation of backup players, RAD52 protein has emerged as a critical gatekeeper of survival in BRCA-deficient cancers. Importantly, RAD52 is largely dispensable in healthy cells but becomes essential for cancer cells with defective BRCA1 or BRCA2 genes. This selective dependency makes RAD52 a coveted drug target for precision oncology, offering a promising new avenue to exploit cancer’s intrinsic vulnerabilities. Targeted inhibition of RAD52 will kill cancer cells with minimal negative effect on healthy cells. Figuring out the functions and features of RAD52 that should be targeted was the challenege. Understanding the structural and functional information about RAD52 will help develop new, specific ways to inhibit this protein.
Our recent study, published in Nature, provides the first insight into an unexpected architecture of how RAD52 interacts with stalled DNA replication forks, a vulnerable structure that forms when replication machinery encounters obstacles. These insights not only deepen our understanding of replication stress but also open new avenues for drug discovery.
Role of Rad52 as a backup mechanism
DNA replication is a high-speed, high-fidelity process. When replication forks stall due to damage or structural impediments, cells deploy specialized DNA repair mechanisms to stabilise and restart them. If mismanaged, stalled forks can collapse, leading to DNA double-strand breaks and genome instability which is a hallmark of cancer.
One common response to fork stalling is fork reversal, where the fork backs up into a “chicken foot” structure. While reversal can be protective, excessive reversal is harmful, causing DNA degradation and cell death. RAD52 prevents this excessive reversal, acting as a molecular shield. But until now, the structural basis of this protection was unknown.
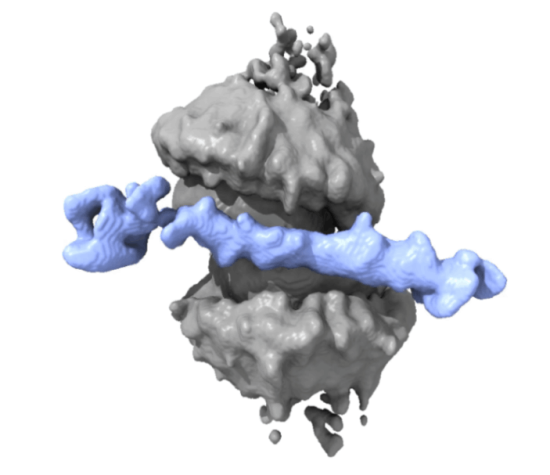
Our discovery: A spool-like double ring
By employing a comprehensive, multi-modal approach including cryo-electron microscopy (cryo-EM), mass photometry, single-molecule imaging, biochemistry, cell-based studies, super-resolution imaging, and computational modelling, we uncovered a novel assembly of RAD52 that is functionally distinct. While prior structural work by Lian and colleagues (2024), Singleton and colleagues (2002) and Kinoshita and colleagues (2023) had established existence of the multiple structures including undecameric 11-subunit and 10-subunit open structure and its role in ssDNA annealing for Homologous Recombination (HR), the functional structure of RAD52 at the stalled replication fork remained unclear.
Our study shows an arrangement that creates a continuous positively charged surface that accommodates both single-stranded and double-stranded DNA regions. The design is not just elegant; it is highly functional. This architecture enables RAD52 to effectively remodel the replication fork into a highly stable configuration. By restructuring the fork, RAD52 creates a resistance mechanism against reversal or collapse mediated by motor proteins like SMARCAL1, ensuring the stability and protection of the fork during stress.
Dynamic remodelling in real time
To visualise this process, we employed single-molecule Förster Resonance Energy Transfer (smFRET), a powerful technique that allows us to measure nanometer-scale distances within individual DNA molecules. By strategically labeling different parts of the replication fork with fluorescent tags (dyes), we could use smFRET to observe how the DNA structure changes or is reorganised upon RAD52 binding. These real-time measurements were captured using a custom-built Total Internal Reflection Fluorescence (TIRF) microscope. This setup enabled us to see the fluorescent signals from single molecules with exceptional clarity, providing direct, molecule-by-molecule evidence of how RAD52 exerts its protective and remodelling effect on the stalled replication fork.
These experiments revealed that RAD52 dynamically rearranges the fork, converting it into a four-way junction through bidirectional strand exchange. Unlike classical recombinases that require ATP for energy, RAD52 operates without energy input, making its mechanism distinct and intriguing.
My role in this work focused on mass photometry analyses. Mass photometry is a method that uses light to “weigh” individual molecules, to determine the precise number of RAD52 units assembling on the DNA. This allowed us to definitively measure that the replication fork forces RAD52 to form a new, heavier complex: a functional double-ring spool made of 22 molecules. We then used mass photometry to show that this specific double-ring complex is what physically displaces and blocks SMARCAL1, proving the double-ring is the active shield.
Competition with SMARCAL1, therapeutic implications
Why does this matter for cancer therapy? Because RAD52’s ability to protect forks hinges on its competition with SMARCAL1, a motor protein that drives fork reversal. Our data show that forming the double-ring structure is key to outcompeting SMARCAL1. Mutations that disrupt RAD52’s outer DNA-binding site abolish this protective function, leading to uncontrolled fork reversal and genome instability.
This mechanistic insight explains why RAD52 inhibition is a potential therapeutic strategy via synthetic lethality. The classic example is the drug class PARP inhibitors, which target the PARP1 enzyme and selectively kill BRCA-mutated cancer cells.
The PARP inhibitors block the PARP1 enzyme, which normally acts as a backup repair protein. When administered in cancer cells with a pre-existing mutation in the BRCA1 or BRCA2 genes, which already lack a primary repair system, this combined defect becomes lethal. This strategy has led to the FDA approval of drugs like Olaparib (Lynparza), Talazoparib (Talzenna), and Rucaparib (Rubraca) for BRCA-mutated cancers.
In the same way, cancer cells with a non-functional BRCA protein are critically dependent on RAD52 for fork protection. Losing RAD52 or inhibiting its function in BRCA-mutated cancer cells triggers catastrophic replication stress, leading to their selective killing.
RAD52 inhibitors already exist in early-stage development, but rational drug design requires detailed structural knowledge. Our study provides that blueprint by mapping critical residues involved in fork engagement, we identify new surfaces for small molecule targeting. Combining RAD52 with PARP inhibitors could overcome resistance and expand treatment options for patients with BRCA-related cancers.
Targeting DNA repair/replication players
While this work focused on RAD52 protein, it aligns with my broader research goal of leveraging mechanistic biochemistry to develop precision cancer therapies. I also study how non-canonical DNA structures, especially G quadruplexes, resolve during replication and how they drive replication stress and therapeutic resistance in cancer cells. These structures formed in guanine-rich regions of the genome. They play important regulatory roles in our cells, but their persistence can stall replication forks, creating DNA breaks and trigger genome instability. A critical gap exists in our understanding of how cells unfold these stable structures to ensure faithful DNA replication.
A turning point
The discovery of RAD52’s double-ring architecture marks a turning point in our understanding of replication fork biology. It illustrates how structural insights can illuminate therapeutic strategies and underscores the power of interdisciplinary approaches to tackle complex biological problems.
As we move forward, the challenge is clear: translate these molecular revelations into clinical solutions. By targeting RAD52 and related pathways, we can expand the arsenal against cancers that currently evade treatment, bringing us closer to the promise of precision medicine.



